
-

邮箱
info@e-omics.com
-

电话
4006321322
-

微信
18515913365
-

QQ
47422642


技术简介
蛋白质组是指在某一特定时间,特定条件下,某一特定组织中表达的所有蛋白质。基于质谱技术的蛋白质组学研究能够在一定范围内检测某一组织中的蛋白质组分和含量。然而许多生物过程是通过分子间的相互作用和化学修饰来调节的,对蛋白质丰度水平变化影响不大,无法使用传统的蛋白质组学进行鉴定。
近年来开发出许多新型的蛋白质组学,例如磷酸化蛋白质组学、互作组学和基于活性的蛋白质组学可以捕获影响蛋白质功能的特定分子事件,但通常报告类型单一,无法进行大范围内对不同的调控事件进行高通量分析。基于此,瑞士苏黎世联邦理工大学生物系分子系统生物学研究所Paola Picotti教授团队开发出一种新型的蛋白质组学技术LiP-MS(1),并在Cell上发表题为“Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ”的文章(2),介绍了LiP-MS技术可在整个蛋白质组范围内监测蛋白质的结构变化,其分辨率可精确定位单个功能位点。
1.1 技术优势
(1) 肽水平的分辨率:结构指纹的改变可以通过~12个氨基酸的分辨率来识别,提示有小分子结合位点。
(2) 能够产生机制假设:检测和区分不同类型的蛋白质-小分子相互作用,包括变构作用、酶-底物、酶-产物和药物-靶标相互作用。
(3) 原位分析:结构变化是在类似自然环境,如细胞裂解液。
(4) 蛋白质组范围:同时测量样本中所有ms可检测蛋白的结构指纹。
(5) 广泛适用性:该方法不仅局限于检测蛋白质-小分子的相互作用,还可用于识别不同类型的蛋白质结构变化的一系列扰动,如温度或生长介质。
1.2 技术应用
Lip-MS是主要开发用于监测蛋白质结构变化、并识别不同条件下结构特异性蛋白水解指纹图谱的新型技术,在此研究中可用于原位监测蛋白功能变化,通过将蛋白质动态结构数据与功能相联系,有助于细胞三维模型的构建,推动结构生物学的进一步发展。
2、Lip-MS实验原理
LiP-MS 检测蛋白构象变化的关键在于 LiP 技术。该技术的核心是让蛋白处于天然构象的条件下,用广谱性的蛋白酶对其进行酶切。首先在非变性的条件下提取蛋白质,保留其原本的空间结构。加入小分子代谢物后,如果该代谢物结合到某些蛋白质上以,就会局部改变蛋白质的结构,进行改变蛋白质被Proteinase K酶切的表位。然后,再使用常规的方法进行蛋白变性和Trypsin酶解,通过质谱分析比较差异的肽段,就可以找到这些被小分子结合的蛋白质。
近年来开发出许多新型的蛋白质组学,例如磷酸化蛋白质组学、互作组学和基于活性的蛋白质组学可以捕获影响蛋白质功能的特定分子事件,但通常报告类型单一,无法进行大范围内对不同的调控事件进行高通量分析。基于此,瑞士苏黎世联邦理工大学生物系分子系统生物学研究所Paola Picotti教授团队开发出一种新型的蛋白质组学技术LiP-MS(1),并在Cell上发表题为“Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ”的文章(2),介绍了LiP-MS技术可在整个蛋白质组范围内监测蛋白质的结构变化,其分辨率可精确定位单个功能位点。
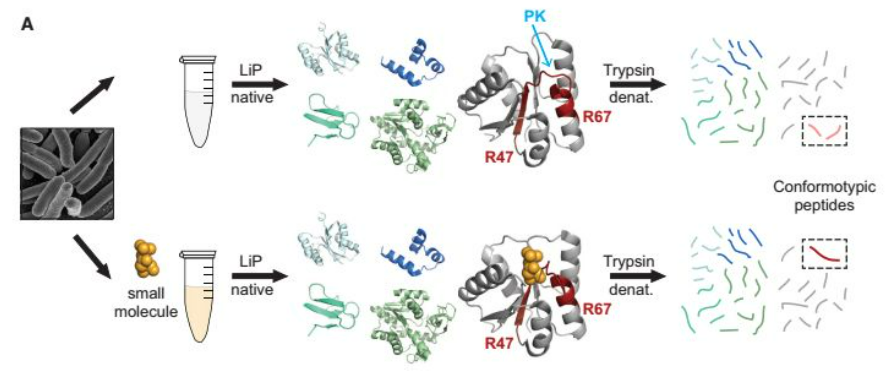
图1 LiP-MS原理示意图
3、Lip-MS实验流程
准备不同处理条件下的样品。酵母、细菌、哺乳动物细胞系、生物液体等样品均可进行lip-ms实验。
3.1非变性条件下蛋白提取
以哺乳动物细胞为例,细胞裂解液可以选择10mM PBS。细胞用PBS漂洗后,加入裂解液,机械力破碎或者反复冷冻破碎。1000g,4℃, 离心 5min,收集上清。(此步骤需全程在冰上进行,裂解液中不能有变性剂。)使用BCA测定法测定蛋白质浓度,并用10mMPBS将蛋白质提取物稀释至1mg/ml。
3.2 酶解:蛋白酶K消化->连续 LysC–胰蛋白酶消化
例如蛋白酶 K(常用)、thermolysin (嗜热菌蛋白酶)、糜蛋白酶(chymotrypsin)、或者弹性水解酶(elastase)。这些蛋白酶倾向于剪切未折叠的蛋白区域。酶与蛋白样品的比例要低,一般为1:100。酶解时间要短,室温酶解 5min,酶解后立即转移至沸水中使蛋白酶灭活。在广谱性蛋白酶作用下,这些非变形的蛋白样品会被切割成大的蛋白片段。
将上述变性的蛋白样品加入脱氧胆酸钠至终浓度5%,按常规流程进行还原烷基化、酶解,即加入 10mM DTT, 55℃,30min,加入40mM IAA, 室温避光反应 30min。样品按酶与底物 1:100 (w/w) 加入LysC蛋白酶37℃,反应4h。样品用100mM NH4CO3将脱氧胆酸钠稀释至1%, 按酶与底物 1:100 (w/w) 加入测序级胰酶,37℃酶解过夜。 停止消化,加入98%(vol/vol)甲酸至最终浓度为2%(vol/vol)沉淀DOC,将样品在室温下以16000g离心10min去除沉淀物。
3.3 多肽脱盐
使用Sep-Pak柱包装10mg C18树脂来去除样品中的盐。使用脱盐缓冲液A(洗涤)和B(洗脱),加载、洗涤和洗脱肽混合物,用真空离心机蒸干多肽溶液,质谱检测。
3.4 质谱检测
LiP得到的肽段样品,根据实验目的,可以依赖多种质谱检测手段进行检测,包括大规模筛选模式(DDA,DIA),或者靶向模式(SRM,PRM)。液相梯度按照常规 2h分离,质谱检测参数按照常规 DDA,DIA, SRM 及 PRM 检测模式设置,需要注意的是质谱检测时电荷数范围选择1-7。搜库软件及参数也同常规,搜库参数中胰酶酶切需要设定为半酶切模式。
3.5 实验要点
(1) LiP 技术必须在非变性条件对蛋白进行酶切,因此只适合与可溶性蛋白,对膜蛋白不适用;
(2) 在进行广谱酶酶切时,需要把握好酶切开始和终止的时间与温度;
(3) 每个条件下蛋白样品均需要设定一个对照组,该对照组仅按照常规蛋白组样品制备流程进行变性、还原、烷基化和胰酶酶解。不同条件下蛋白构象的变化,在两步酶切中,会产生不同酶切形式的肽段,通过比较肽段的不同酶切模式,可以研究蛋白构象的变化。
4、LiP-MS数据分析
首先根据LC-MS/MS检测结果,对蛋白质和多肽进行鉴定定量分析,然后筛选差异多肽,并对差异多肽中感兴趣的目标多肽进行构象可视化分析。
4.1 蛋白质及多肽的定量分析
(1) 将不同处理的MS原始数据导入Proteome Discoverer,使用搜索引擎工具对谱图进行数据库搜索,对每个样品的肽离子图谱进行比对和谱峰量化处理。
使用参数设置如下:
母体和片段质量容差为10 ppm,
胰蛋白酶(对照样品选择全酶切,LiP-MS样品选择半酶切),
特异性胰蛋白酶 (半特异性)的最大漏切位点为2,
半胱氨酸氨基甲基化 (carbamidomethylation on cysteines) 作为固定修饰,
蛋氨酸氧化 (oxidation of methionines) 作为可变修饰。
(2) 分析仅用胰蛋白酶处理的对照样品,对蛋白质进行定量分析。
(3) 分析LiP样品,对多肽进行定量分析。
4.2 差异多肽的筛选
(1) 计算多肽丰度的变化倍数。
(2) 计算蛋白质丰度的变化倍数。
(3) 使用蛋白质丰度的变化倍数作为多肽水平LiP数据的归一化因子。这是通过将不同样品的肽水平丰度变化除以各自蛋白质的丰度变化来实现的。对于未显著改变丰度的蛋白质,使用1作为标准化因子。
(4) 使用适当的阈值筛选差异多肽。建议使用|log2FC| > 1和p < 0.01作为阈值。LiP样本中丰度显著发生变化的多肽,被认为位于结构发生变化的蛋白质特定区域。
(5) 以火山图的形式展示多肽丰度的变化倍数与相关q值的关系。
4.3 目标多肽构象的可视化
将筛选得到的差异多肽映射到对应的蛋白质3D结构上,以评估蛋白质功能与结构的相关性。
(1) 确定目标多肽及相应蛋白质在PDB数据库中的ID;
(2) 使用在线工具将目标多肽匹配到蛋白质的3D结构上。
5、文献研究实例
5.1 实例1
分析不同应激条件下蛋白质结构变化
许多生物过程是通过分子间的相互作用和化学修饰来调节的,对蛋白质丰度水平变化影响不大,无法使用传统的蛋白质组学进行鉴定。Paola Picotti团队利用开发出的新型蛋白质组学技术LiP-MS,实现了在整个蛋白质组范围内监测蛋白质的结构变化,其分辨率可精确定位单个功能位点(2)。
作者首先通过对处于指数生长期的酵母施加渗透胁迫或热胁迫,随后提取蛋白质组进行LiP-MS实验,同时对蛋白质的丰度和结构变化进行测定。结果发现,短时间刺激下丰度发生变化的蛋白质仅占1%,而结构发生变化的蛋白质在热激和渗透压刺激下分别为23%和11%。为了验证这些结构发生变化的蛋白质是否是由刺激响应引起的,作者对这些蛋白质进行功能富集分析,与之前研究结果一致,例如发现了与翻译调控和蛋白质折叠、错误折叠和复性相关功能途径富集于热应激数据集。
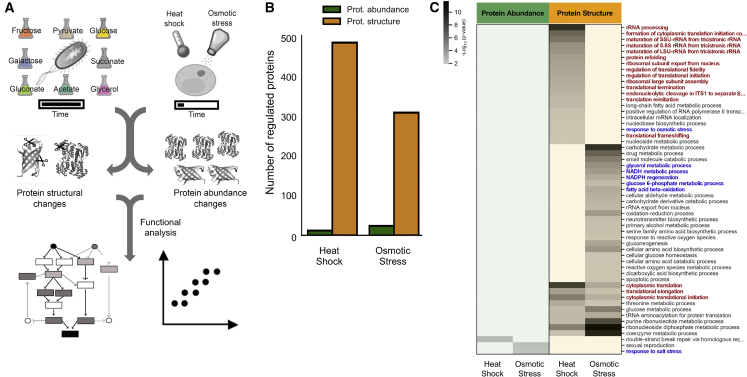
图2 酵母和大肠杆菌细胞反应过程中整体蛋白结构和丰度的变化
作者通过对结构发生改变的蛋白质进行分析发现,它们和已报导过的酵母对渗透压胁迫的途径类似,同时找出HOG-MAPK代谢途径中12种在渗透应激条件下结构发生明显改变的激酶或磷酸酶(下图中用橘色表示)。
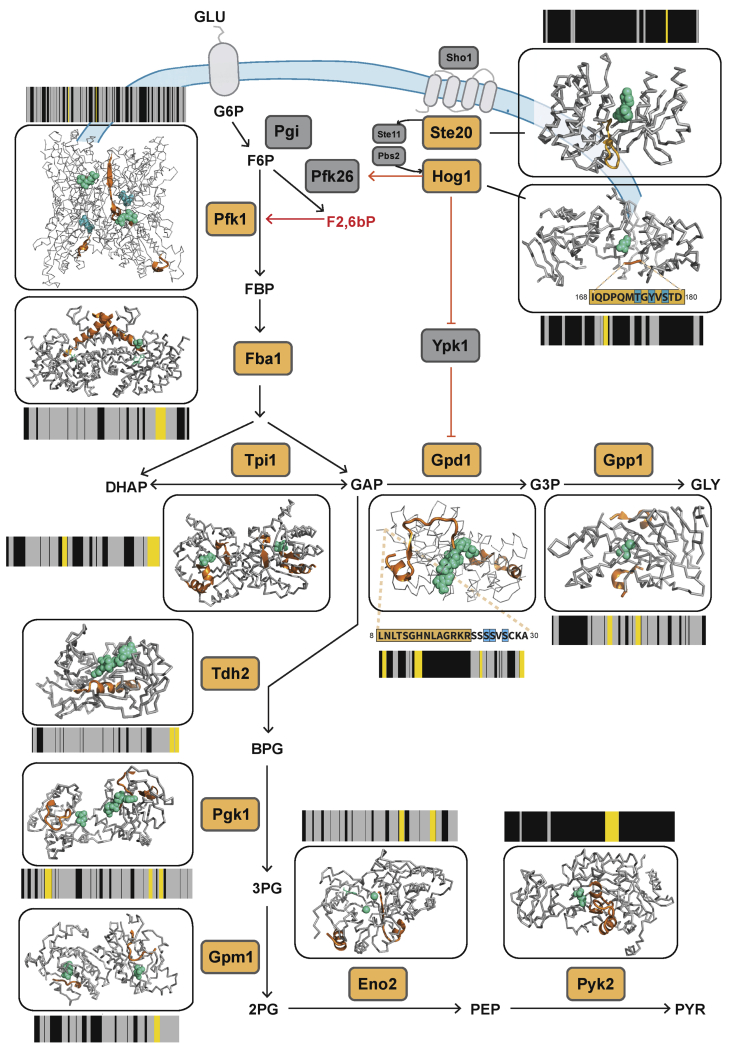
图3 结构变化捕获酵母应对渗透休克时的多种调节事件
作者又用Lip-MS方法研究了在8种不同碳源中生长的大肠杆菌。在相同的生长条件下,代谢产物水平通常和大肠杆菌中央碳代谢(CCM)的通量表现出条件依赖性。作者认为流量变化可以用来评估酶功能状态的改变,并以此报告CCM的功能变化。与葡萄糖相比,每种生长条件下蛋白质结构和丰度的差异分析表明,平均共有365种蛋白质发生了结构改变,190种蛋白质发生了丰度变化,正如在酵母实验中所观察到的,更多的蛋白质发生的是结构改变。
类似地,作者也对不同碳源中结构和丰度变化的蛋白质进行功能富集分析。结果显示,在丰度改变的蛋白质中不存在糖酵解途径的富集,这与先前的观察一致,即糖酵解主要不是在转录水平受到调控,与此相反,在所有生长条件下发生结构改变的蛋白质中均存在糖酵解途径的富集。这些结果提示,不同的调控机制控制着生长在不同碳源上的大肠杆菌代谢途径,其中糖酵解主要受蛋白质结构而非基因表达的调控,而三羧酸循环和乙醛酸循环除了影响蛋白质结构的影响,还受到蛋白质丰度的调节。
对于特定的酶促反应,反应通量的变化可能是由于酶活性、底物浓度或酶含量变化造成的。作者推测酶活性改变或与反应物结合影响蛋白质结构而产生LiP信号。通过线性回归测试研究在8种条件下酶的结构变化或丰度变化与代谢通量变化的相关性。基于此,作者假设代谢物FBP可与大肠杆菌ptsl酶的活性位点结合。首先,通过计算分析表明FBP可能是ptsl的竞争性抑制剂,而体外实验证实FBP降低ptsI活性。先前研究表明ptsI控制己糖摄取并调节糖酵解流量,有趣的是,FBP被证明是一种细胞内糖酵解流量传感器,因此,作者假设FBP-ptsI相互作用是一个负反馈回路,在糖酵解中间产物已经很丰富的情况下,可以防止过量的葡萄糖摄取,随后作者也通过实验验证这一观点。
生物过程是由分子间相互作用和化学修饰来调节的,对蛋白质表达水平影响不大,传统蛋白质组学无法检测出其中的变化。LiP-MS技术可以通过对蛋白质结构变化原位检测蛋白质功能变化,将动态和高分辨率的结构数据与功能联系起来,使得细胞功能的3D模型构建更加完善。
5.2 实例2
识别和预测靶蛋白
药物与靶蛋白结合可以改变蛋白质的酶解可及性,从而改变非限制性酶切模式。通过LiP-MS可以识别和预测药物的靶向激酶。
Paola Picotti等人利用开发的LiP-Quant方法对靶向激酶和磷酸酶的化合物库进行了方法验证,并在全细胞裂解液中预估化合物结合位点的EC50值,筛选出选择性强的靶蛋白(3)。
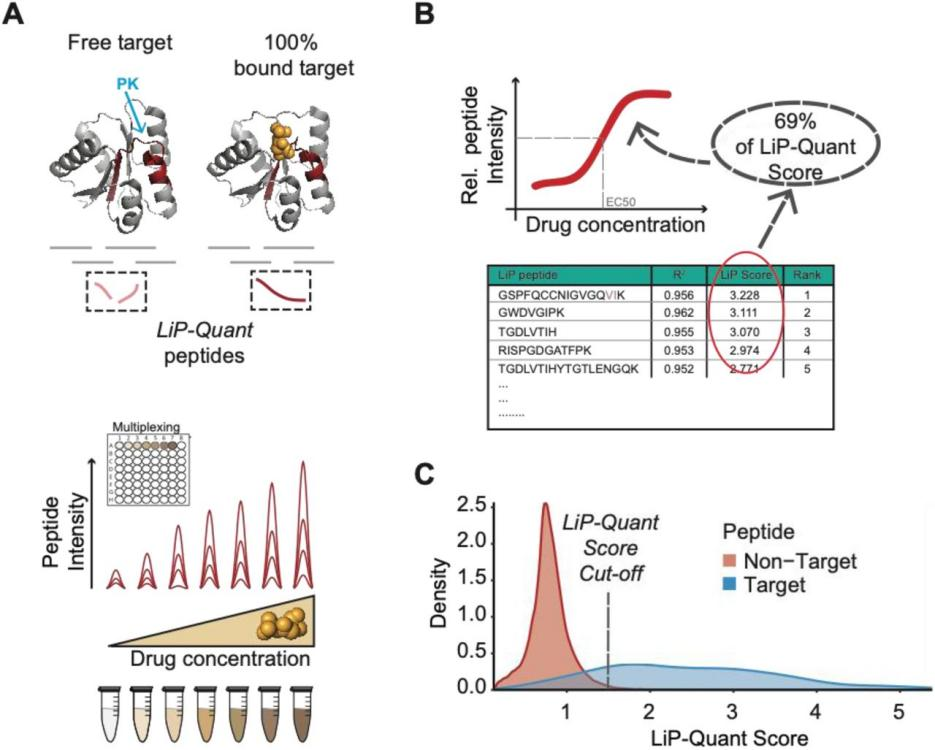
图4 LiP-Quant方法的流程图
靶蛋白中结合位点的酶切特征会由于给药而改变,真正的靶肽段会随着药物剂量丰度随之变化;图4B为作者建立的一套LiP-Quant综合得分体系,以此来更好地筛选真正的靶蛋白;图4C显示LiP-Quant得分呈现出双峰分布,已知靶蛋白的肽段明显富集在高得分的峰中(得分大于1.5),而非靶点的肽段多富集在低分区,根据此结果,作者定义得分大于1.5分的肽段为LiP-Quant肽段。
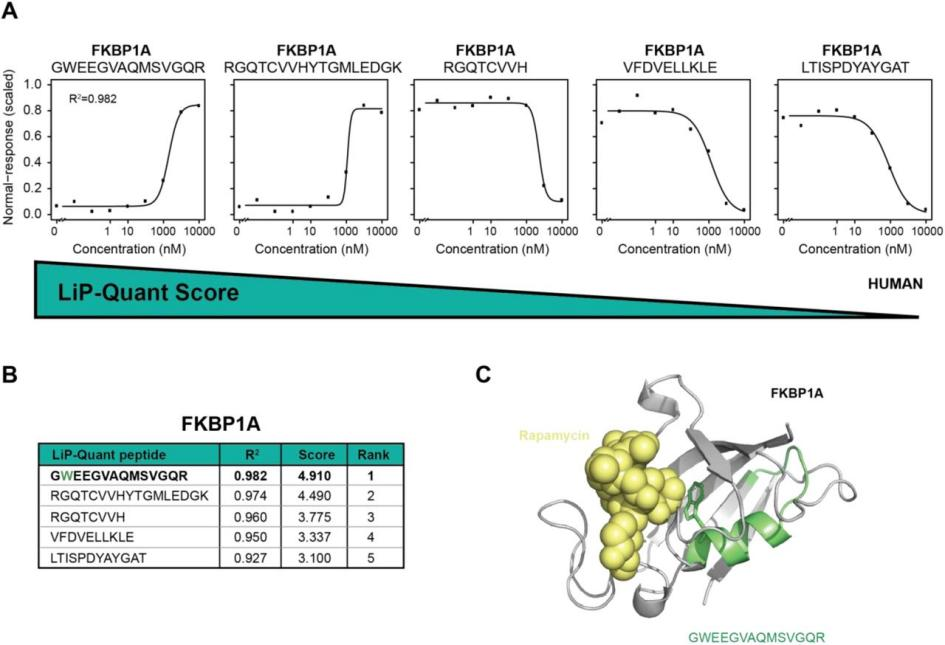
图5 人类细胞中LiP-Quant测试
A,LiP-Quant肽段的剂量反应测试曲线;
B,雷帕霉素实验中LiP-Quant肽段的得分排序;
C,FKBP1A与雷帕霉素复合物的结构模型。
随后,作者对靶点明确的免疫抑制药物雷帕霉素分别在酿酒酵母和Hela细胞中进行测试,结果发现包括排名Top5在内的多个LiP-Quant肽段对应雷帕霉素在酿酒酵母中的靶标FRP1蛋白和Hela细胞中的FKBP1A蛋白,从而证明了LiP-Quant在酵母和人类细胞中鉴定药物靶点的可行性。两个实验中得分Top2的肽段对应的正是雷帕霉素和靶蛋白已知的结合位点(图5C)。

图6 用LiP-Quant筛选药物靶标
A,具有不同特异性的药物靶标的LiP-Quant分数分布情况;
B,雷达图显示不同阈值下LiP-Quant肽的比例;
C,不同LiP-Quant肽段的剂量反应曲线;
D,MAP2K1及其LiP-Quant肽的三维构象。
接下来作者聚焦在靶向激酶和磷酸酶的药物上。作者选择了常用的广谱激酶抑制剂staurosporine和人类细胞系中特异性抑制MAP激酶的selumetinib做测试。对于staurosporine,LiP-Quant得分Top25肽中有18个肽对应13种蛋白激酶而selumetinib则只测到来自MAP2K1/2的肽段,这意味着LiP-Quant方法可以完成选择性和广谱化合物的靶标筛选(图6A、6B)。在两个药物靶蛋白筛选中NQO2都有着很高的得分(图6C),这可能代表此蛋白是这两者的常见脱靶蛋白。LiP方法的独特优点在于它具有肽水平发现相互作用的能力,在星形孢菌素和司美替尼实验中LiP-Quant方法鉴定出的LiP-Quant肽的位置都与已知激酶抑制剂结合位点非常接近,甚至在司美替尼实验中能找出MAP2K1的异构位点(图6D)。
5.3 实例3
探讨蛋白质药物的临床作用机制
通过LiP-MS可以获得药物与蛋白质的具体结合位置信息,从而进一步分析它们的相互作用机制。RIPK1是一种含死亡结构域的激酶,被认为是病理条件下抑制凋亡、坏死和炎症的重要治疗靶点,RIPK1激酶抑制剂已被用于治疗各种人类疾病的临床研究。目前在研发RIPK1抑制剂的瓶颈之一是发现新的方法来抑制这种激酶。Huyan Meng等人发现Necrostatin-34(Nec-34)抑制RIPK1激酶的机制不同于已知的RIPK1抑制剂(如 Nec-1)。机制研究表明,Nec-34通过在激酶结构域中占据一个独特的结合口袋来稳定RIPK1激酶的非活性构象。此外,研究团队还发现体内体外实验中Nec-34系列化合物均可协同Nec-1s抑制RIPK1的活性。因此,Nec-34提供了靶向RIPK1激酶的新策略,并为RIPK1介导的疾病提供了潜在的组合治疗方案。
研究人员使用LiP-MS进一步验证了Nec-34的结合模式(4)。与非结合对照组中的LiP模式相比,用Nec-34处理特异性增加了肽133-GVIHKDLKPENIL-145和173-HNELREVDGTA-183的丰度(图a及图c,蓝色肽) ,它们位于计算预测的484结合口袋中(图d)。
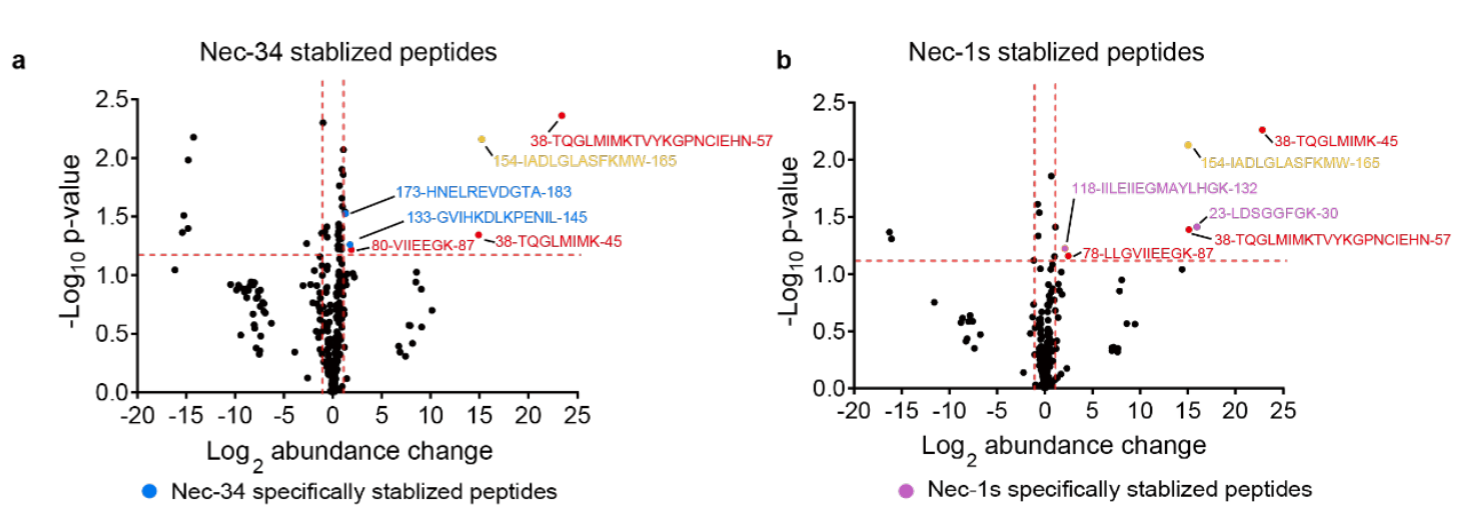
图7 用火山图展示LiP-MS鉴定到的肽
相比之下,用Nec-1s处理导致肽118-IILEIIEGMAYLHG-131和22-ELDSGGFGK-30的丰度增加(图8b及图8c,洋红色的肽)。Nec-1s和Nec-34的结合都增加了154-IADLGLASFKMW-165肽的丰度,其位于X射线共结晶结构中Nec-1s的结合口袋17和预测的484结合口袋之间(图7a、图7b及图7c,黄色肽)。
位于Nec-34和Nec-1s结合口袋远端区域的两种肽38-TQGLMIMK-45和78-LLGVIIEEGK-87均能为Nec-34和Nec-1s所稳定(图7a、图7b及图7c,红色肽) ,这表明尽管Nec-34及其类似物484和496占据不同于Nec-1s的结合口袋,但它们可能能够诱导RIPK1激酶中类似的抑制构象变化。
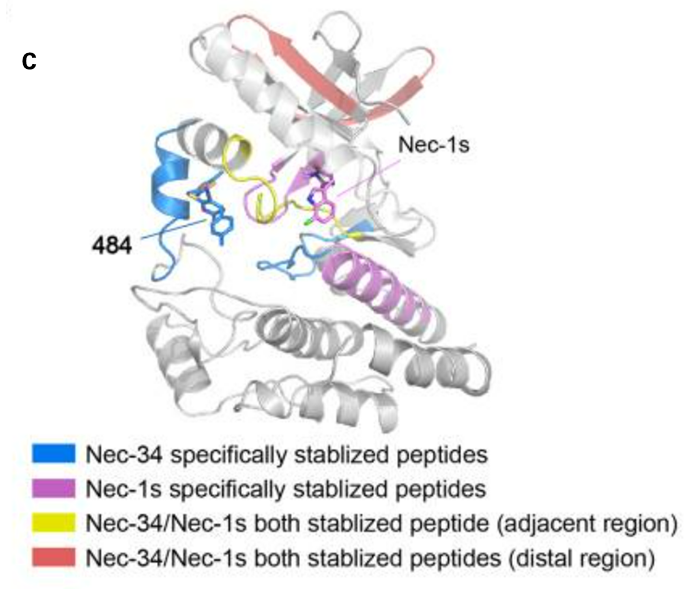
图8 确认RIPK1激酶中的Nec-34结合位点
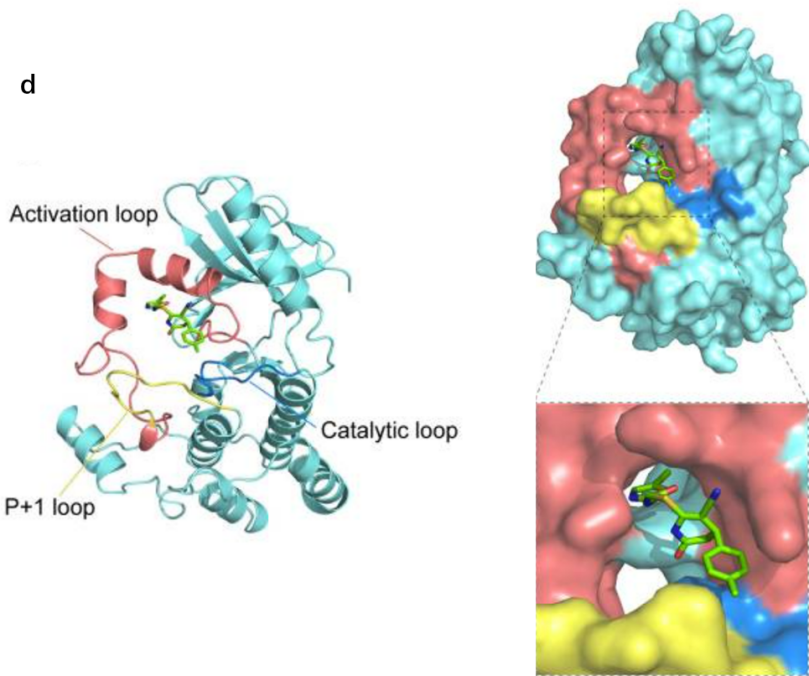
图9 识别RIPK1激酶中的Nec-34结合区域
参考文献
1. S. Schopper et al., Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nature Protocols 12, 2391-2410 (2017).
2. V. Cappelletti, T. Hauser, I. Piazza, M. Pepelnjak, P. Picotti, Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ ll Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ. Cell, 1-15 (2021).
3. Y. Feng et al., LiP-Quant, an automated chemoproteomic approach to identify drug targets in complex proteomes - ScienceDirect. European Journal of Cancer 138, (2020).
4. H. Meng et al., Discovery of a cooperative mode of inhibiting RIPK1 kinase. Cell Discov 7, 41 (2021).
